What is phylogenetics?
Phylogenetics is the study of evolutionary relationships among biological entities - often species, individuals or genes, which may be referred to as taxa [1]. Phylogenetic trees can be constructed by analyzing nucleotides or protein sequences combined with an understanding of sequence evolution. One would then be able to infer evolutionary events that occurred in the past as well as have more information about the evolutionary processes operating on sequences.
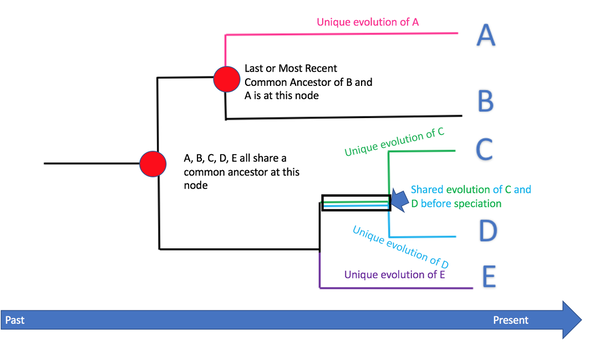
Figure 1. Phylogenetic Tree Diagram
Methods for Constructing Phylogenetic Trees
Maximum Likelihood [2]: This method recreates the series of nucleotide changes that resulted in the pattern of variation revealed by a multiple alignment. The assumption is that evolution follows the shortest possible route and that the correct phylogenetic tree is therefore the one that requires the minimum number of nucleotide changes to produce the observed differences between the sequences. Trees are therefore constructed at random and the number of nucleotide changes that are calculated until all possible topologies have been examined, and the one requiring the smallest number of steps identified. This is presented as the most likely inferred tree.
Neighbor Joining [3]: This method is a popular tree-building procedure that uses a distance matrix approach. To begin the tree construction, it is initially assumed that there is just one central node from which branches leading to all the DNA sequences radiate in a star-like pattern. Next, a pair of sequences is chosen at random, and the distance matrix is used to calculate the total branch length in this new ‘tree’. This operation is repeated until all the possible pairs have been examined enabling the combination that gives the tree with the shortest total branch length to be identified. This pair of sequences will be neighbors in the final tree.
Minimum Evolution [2]: This method creates scores based on sequence similarities between two species. Species most closely related will have equal branch lengths and diverge an equal distance from their common ancestor. To build this phylogenetic tree takes more time than would building a tree via the neighbor joining method.
Neighbor Joining [3]: This method is a popular tree-building procedure that uses a distance matrix approach. To begin the tree construction, it is initially assumed that there is just one central node from which branches leading to all the DNA sequences radiate in a star-like pattern. Next, a pair of sequences is chosen at random, and the distance matrix is used to calculate the total branch length in this new ‘tree’. This operation is repeated until all the possible pairs have been examined enabling the combination that gives the tree with the shortest total branch length to be identified. This pair of sequences will be neighbors in the final tree.
Minimum Evolution [2]: This method creates scores based on sequence similarities between two species. Species most closely related will have equal branch lengths and diverge an equal distance from their common ancestor. To build this phylogenetic tree takes more time than would building a tree via the neighbor joining method.
CLN3 Phylogenetic Tree Construction
Step 1: Obtain protein FASTA sequences from the organisms of interest. Put the protein homologs in a .txt file.
Step 2: Sequences must then be aligned (can be done via Clustal Omega or MEGA). The alignment below was conducted using MEGA.
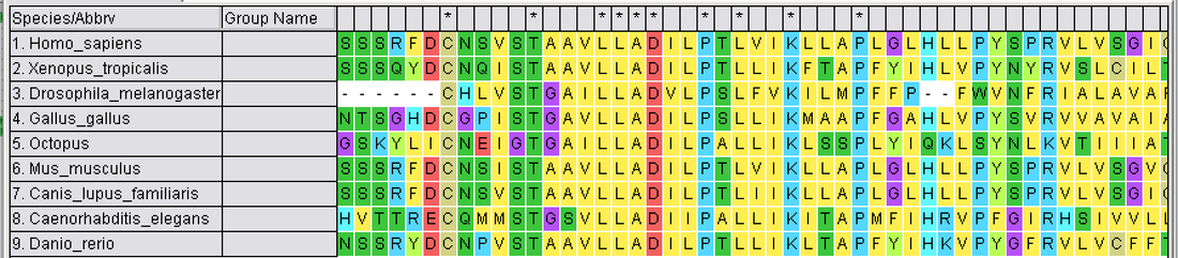
Figure 2. Sequence alignment of CLN3 homologs in MEGA
Step 3: Within the MEGA program, phylogenetic trees can be created using Maximum Likelihood, Neighbor Joining, and Minimum Evolution. All three trees are listed below
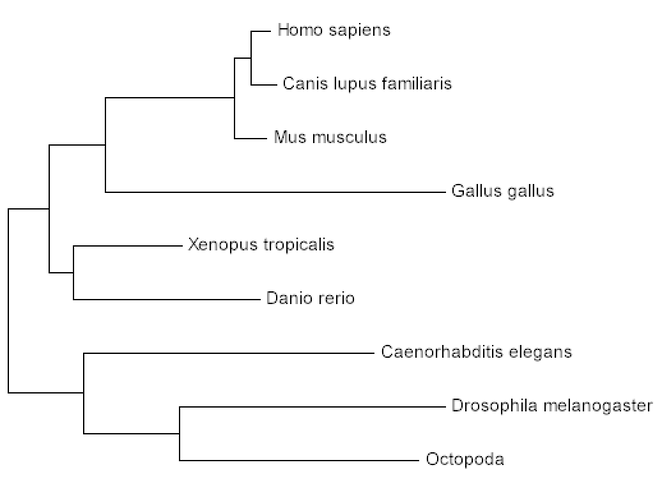
Figure 3. Phylogenetic tree of CLN3 homologs via Maximum Likelihood
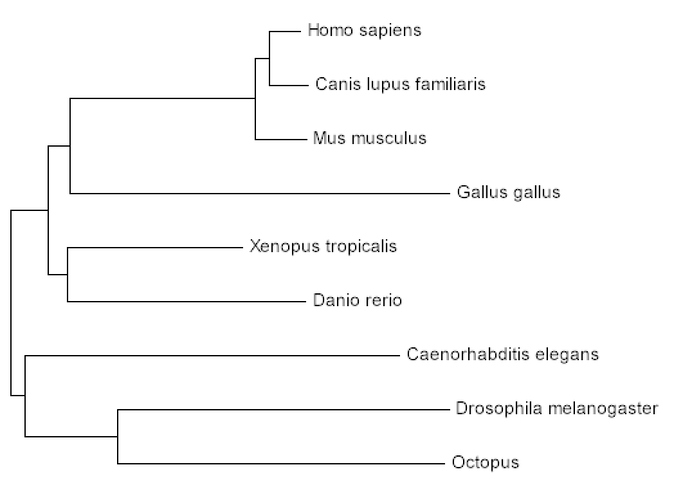
Figure 3. Phylogenetic tree of CLN3 homologs via Neighbor Joining
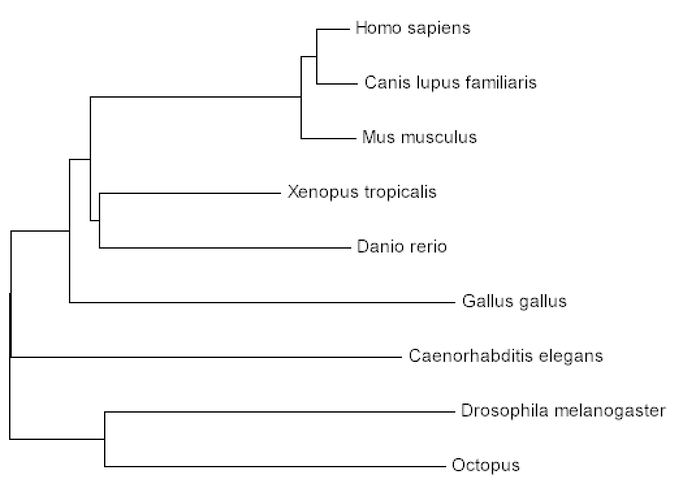
Figure 3. Phylogenetic tree of CLN3 homologs via Minimum Evolution
Conclusion:
The use of phylogenetics grants us the opportunity to understand the prevalence of CLN3 throughout different homologous organisms. In addition, the percent identities that were found for each homolog through BLAST were verified in the creation of these trees. Percent identities don't always reflect the evolutionary pattern of a given gene. This is the case for CLN3 given the phylogenetic trees display that the chicken (Gallus gallus; 78% identity) is more closely related to humans than the fruit fly (Drosophila melanogaster; 92% identity), octopus (Octopoda; 92% identity), and the nematode (C. elegans; 91% identity). The phylogenetic trees, however, are more accurate for evolutionary patterns, thus these trees can be used to visualize the evolutionary pattern of CLN3.
References:
[1] What is phylogenetics? (2016, June 08). Retrieved from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[2] Phylogenetic Reconstruction. (n.d.). Retrieved from http://evolution-textbook.org/content/free/contents/ch27.html#ch27-4-4
[3]Brown, T. A. (1970, January 01). Molecular Phylogenetics. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK21122/
Header: http://www.technocrazed.com/tree-wallpapers-backgrounds-hd-free-download
Figure 1: https://socratic.org/questions/how-are-phylogenetic-trees-read
[2] Phylogenetic Reconstruction. (n.d.). Retrieved from http://evolution-textbook.org/content/free/contents/ch27.html#ch27-4-4
[3]Brown, T. A. (1970, January 01). Molecular Phylogenetics. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK21122/
Header: http://www.technocrazed.com/tree-wallpapers-backgrounds-hd-free-download
Figure 1: https://socratic.org/questions/how-are-phylogenetic-trees-read
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
